- 952
- ۱۴۰۲/۰۲/۰۸ - ۱۱:۲۵
- 845 بازدید
شرح فصل و نکات ویژه: در این فصل به معرفی الگوریتمها و ابزارهایی که میتوان در پیشگویی ساختاردوم و سوم پروتئینها استفاده کرد میپردازیم. همواره پیشگویی با خطاهایی روبرو است. اما مسئله مهم این است که اگر اطلاعاتی از ساختار پروتئین مورد نظر داشته باشیم بهتر از این است که در مورد ساختار آن چیزی ندانیم. تلاش برای درک ساختار پروتئینها باعث شده است تا روشها و ابزارهای پیشگویی مداوم به روز شوند و ابزارهای قویتری در[…]
شرح فصل و نکات ویژه:
- در این فصل به معرفی الگوریتمها و ابزارهایی که میتوان در پیشگویی ساختاردوم و سوم پروتئینها استفاده کرد میپردازیم.
- همواره پیشگویی با خطاهایی روبرو است. اما مسئله مهم این است که اگر اطلاعاتی از ساختار پروتئین مورد نظر داشته باشیم بهتر از این است که در مورد ساختار آن چیزی ندانیم. تلاش برای درک ساختار پروتئینها باعث شده است تا روشها و ابزارهای پیشگویی مداوم به روز شوند و ابزارهای قویتری در اختیار محققین قرار گیرد.
۲۴۸-فصل دوازدهم
پروتئینها ماکرومولکولهایی هستند که تمام اعمال مهم در ارگانیسمها را انجام میدهند از جمله این اعمال انجام واکنشهای بیوشیمیایی، انتقال مواد غذایی و تشخیص و انتقال پیامها است. بنابراین ژنها خزانه اطلاعات و پروتئینها ماشینهای حیات میباشند. پروتئینها از بهم پیوستن اسیدهای آمینه توسط پیوندهای پپتیدی به صورت یک زنجیره بوجود میآیند. پروتئینها از نظر طول زنجیره (از ۳۰ تا بیش از ۰۰۰/۳۰ اسید آمینه) و همچنین ترتیب قرار گرفتن اسیدهای آمینه، با هم متفاوت هستند. پروتئین پیش از بدست آوردن ساختار سه بعدی نهایی از یک ساختار منظم به نام ساختار دوم گذر میکند.
پروتئینهایی که ما در طبیعت مشاهده میکنیم از طریق انتخاب طبیعی تکامل یافته اند، تا عملی خاص را انجام دهند. عملکرد پروتئینها به ساختارفضایی آنها بستگی دارد. اسیدهای آمینه که با ترتیبی خاص کنار هم قرار گرفته اند (ساختار اول) با پیچ و تاب[۱] خوردن در رو و کنار یکدیگر زیر واحدهایی را میسازند (ساختار دوم) که با استفاده از آنها ساختار فضایی پروتئین ساخته میشود (ساختار سوم) و حتی گاه چند واحد اینچنینی با قرار گرفتن در کنار هم ساختمان عظیمتری (ساختمان چهارم) را بوجود میآورند. توالی آمینواسیدی یا ساختار اولیه یک پروتئین را نخستین بار فردریک سانگر ۱۹۵۳ برای انسولین تعیین کرد. ساختار سه بعدی یا ساختار سوم را نخستین بار جان کندرو در ۱۹۶۰ با استفاده از پراش پرتو x بلورهای پروتئین برای میوگلوبین شرح داد و همچنین روش پراش اشعه x را ماکس پروتز تکمیل کرد.
اگر هر آنچه از اهمیت و عملکرد پروتئینها میدانیم را وابسته به ساختمان فضایی آن بدانیم، برای درک فعالیتهای زیستی پروتئینها علاقمندیم که قادر به شناختن یا پیشگویی ساختمان سه بعدی از روی توالی اسیدهای آمینه باشیم. با وجود تلاشهای قابل ملاحظه در طول سالهای گذشته هنوز این امر میسر نیست و مسأله چگونگی تاخوردن توالی اسیدهای آمینه به ساختمان فضایی پروتئین حل نشده باقی مانده است.
۱-۱۲ ساختمان پروتئینها
ساختمان پروتئینها بطور تجربی بوسیله تکنیکهای بلور نگاری[۲] پرتوی ایکس و رزنانس مغناطیسی هسته[۳] تعیین میشود. در طول ۳۰ سال گذشته ساختمان بیش از ۱۴۰۰۰ پروتئین شناخته شده است و توالی بیش از ۶۰۰۰۰۰ پروتئین مشخص شده است. این موضوع حجم زیادی از اطلاعات را فراهم آورده است که میتوان با استفاده از آنها اصول بنیادی ساختمان پروتئینها را استنتاج کرد.
این اصول درک چگونگی ایجاد ساختمان پروتئینها، رابطه ساختمان و عمل و اساس ارتباط خویشاوندی بین پروتئینها را آسان کرده است. علم ساختمان پروتئین در مرحله شناسایی و طبقه بندی است که در این مرحله ما میتوانیم اجزای شاخص و الگوها را در پروتئینهایی که ساختمان سه بعدی آنها تعیین شده است مشخص کنیم.
در زنجیر اسیدهای آمینه ی موجود در یک توالی سه نوع پیوند وجود دارد که در طول زنجیره تکرار میشود. این سه نوع پیوند به شرح زیر میباشند:
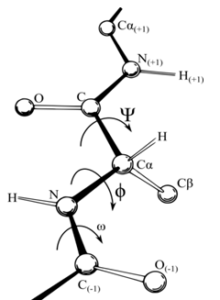

پیوند ω: پیوند بین گروه آمین یک اسید آمینه با گروه کربوکسیلیک اسید آمینه مجاورش را پیوند امگا گویند. این پیوند دارای رزونانس و خصوصیات یک پیوند دوگانه است و از آنجا که پیوند دوگانه توانایی چرخش را ندارد این پیوند نمیتواند هر زاویه پیوندی را ایجاد کند.
پیوند ψ: پیوند بین کربن مرکزی هر اسید آمینه (کربن الفا) با گروه کربوکسیلیک مجاورش را زاویه سای گویند.
پیوند Φ :پیوند بین کربن مرکزی هر اسید آمینه (کربن آلفا) با گروه آمین مجاورش را زاویه فی گویند.
اولین نتایج ساختمانی بلور نگاری روی پروتئینهای کروی در ۱۹۵۸ برای میوگلوبین گزارش شد و مانند یک شوک به کسانی که امیدوار بودند پروتئین ساختمانی ساده داشته باشد و دارای اصول عمومی شبیه به ساختمان ساده DNA دو رشته ای باشد، وارد شد. جان کندرو [۴]پس از تعیین ساختمان میوگلوبین این ناامیدی در مورد پیچیدگی ساختمان پروتئین را با جملات زیر بیان کرد : “شاید قابل توجه ترین ویژگی ملکول، پیچیدگی آن و فقدان تقارن باشد که از هر آنچه که بوسیله تئوری ساختمان پروتئین پیش بینی میشد پیچیده تر است.”
امروزه روشن است که این بی نظمی برای پروتئینها – بدلیل اعمال بسیار متنوعشان – لازم است. ذخیره اطلاعات و انتقال آنها از DNA ضرورتاً خطی است و ملکولهای DNA با محتوای اطلاعاتی مختلف ساختمان کلی یکسانی دارند.
[۱] Folding
[۲] Crystallography
[۳] NMR
[۴] John Kendrew
۲۴۹-پیشگویی ساختار دوم و سوم پروتئین
بر عکس پروتئینها باید بوسیله هزاران ملکول متفاوت در سلول بوسیله میانکنشهای ساختمان سوم تشخیص داده شوند که این موضوع مستلزم آنست که ساختمان پروتئینها متنوع و غیر منظم (فاقد نظمی ساده) باشد. علی رغم این نیاز ویژگیهای منظمی در ساختمان پروتئینها وجود دارد که مهم ترینشان ساختمان دوم آنها میباشد.
مارپیچ آلفا از عناصر کلاسیک ساختمان پروتئین است که اولین بار در ۱۹۵۱ توسط پائولینگ[۱] توصیف شد. او پیشگویی کرد که این ساختمان باید پایدار و از نظر انرژتیک در پروتئین مناسب باشد. مارپیچ آلفا در پروتئین وقتی پیدا میشود که دریک قطعه از توالی، همگی زوایای φ و ψ تقریباً ۶۰- و ۵۰- داشته باشند. در مارپیچ آلفا در هر دور ۶/۳ واحد اسیدآمینه دارد با پیوندهای هیدورژنی بین گروه کربونیل باقیمانده n وگروه آمینو باقیمانده n+4. بنابراین همه گروههای آمینو و کربونیل به استثنای اولین آمینو و آخرین کربونیل در انتهاهای مارپیچ با هم پیوند هیدروژنی دارند. در نتیجه انتهاهای مارپیچ آلفا قطبی هستند و اغلب در سطح پروتئین یافت میشوند.
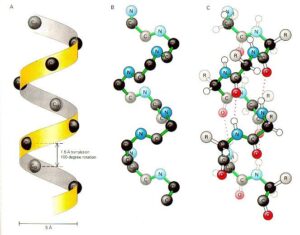
تصویر ۲-۱۲: مارپیچ آلفا. اتمهای اکسیژن و نیتروژن زنجیره اصلی در مارپیچ آلفا با یکدیگر پیوند هیدروژنی دارند.
مارپیچهای آلفا که در پروتئینها مشاهده میشوند اغلب راستگرد هستند. گاهگاهی قطعات کوچک (۳ تا ۵ باقیمانده) از مارپیچهای آلفا چپگرد مشاهده میشود. همه پیوندهای هیدروژنی در یک مارپیچ آلفا در یک جهت هستند به دلیل آنکه واحدهای پپتیدی در یک جهت در طول محور مارپیچ دنبال هم قرار گرفتهاند.
اثر کلی یک دو قطبی خالص برای مارپیچهای آلفا این است که یک بار مثبت جزئی درانتهای آمینو و بار منفی جزئی در انتهای کربوکسی مارپیچ آلفا دارد این بار برای جذب لیگاندهای با بار مخالف و لیگاندهای با بار منفی بهویژه آنهایی که دارای گروه فسفات هستند و معمولاُ به انتهای نیتروژن مارپیچ آلفا وصل میشوند اثر دارد.
دیده شده است که زنجیرههای جانبی مختلف تمایل[۲] ضعیف اما معینی برای حضور در مارپیچ آلفا یا عدم حضور در آن دارند. بنابر این آلانین، گلوتامیک اسید، لوسین و متیونین تشکیل دهندههای خوب مارپیچ آلفا هستند در حالیکه پرولین، گلایسین، تایروزین و سرین از این نظر بسیار ضعیف هستند.چنین تمایلاتی برای هر تلاش اولیه برای پیشگویی ساختمان دوم از توالی اسیدهای آمینه نقطه مرکزی هستند اما به اندازه کافی قوی نیستند که باعث یک پیشگویی دقیق شوند.
[۱] Pauling
[۲] preference
۲۵۰-فصل دوازدهم
عمومیترین مکان برای مارپیچهای آلفا در ساختمان پروتئین در طول سطح خارجی پروتئین است که یک سمت از مارپیچ به طرف حلال و سمت دیگر به طرف آبگریز داخلی پروتئین است. بنابراین با ۶/۳ اسیدآمینه در هر دور تمایلی برای زنجیرههای جانبی به تغییر از آبگریز به آبدوست با تناوب سه یا چهار اسید آمینه[۱] وجود دارد. هر چند این گرایش بعضی مواقع در توالی اسیدهای آمینه دیده میشود اما به اندازه کافی برای پیشگویی ساختمان به تنهایی کافی نیست. زیرا اسید آمینههایی که به سمت محلول هستند میتوانند آبگریز باشند و به علاوه مارپیچهای آلفا میتوانند بطورکامل در داخل پروتئین یا کاملاً در معرض حلال باشند. همانطور که در شکل زیر مشاهده میکنید انواع مختلفی از هلیکس وجود دارد.آلفا هلیکسی که به صورت معمول میشناسیم ۴.۱۳ هلیکس میباشد، یک هلیکس دیگر به نام ۳.۱۰ هلیکس نیز وجود دارد که نازکتر و بلندتر از آلفا هلیکس معمول میباشد.
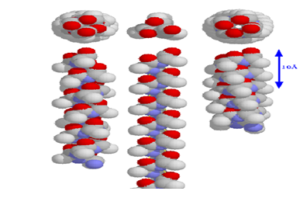
تصویر ۳-۱۲: انواع مارپیچ آلفا. سمت راست pi helix، وسط ۳.۱۰ helix و سمت چپ alpha helix.
دومین عنصر ساختمانی اصلی که در پروتئینهای کروی پیدا میشود صفحات بتا هستند. این ساختمان از ترکیب چندین منطقه زنجیر پلی پپتیدی ساخته شده است – بر خلاف مارپیچ آلفا که از یک منطقه پیوسته ساخته شده است. این مناطق، رشتههای بتا، معمولاً ۵ تا ۱۰ اسید آمینه طول دارند. این رشتههای بتا در مجاور یکدیگر ردیف میشوند (شکلهای ۵ و۶) بهطوریکه پیوندهای هیدروژنی میتوانند بین گروههای کربونیل یک رشته بتا و گروههای آمین روی رشته مجاور و بر عکس تشکیل شوند. صفحات بتا که از چندین رشته بتا تشکیل میشوند چین خورده[۲] هستند و اتمهای کربن آلفا به ترتیب بالا و پایین صفحه صفحات بتا وجود دارند. زنجیرههای جانبی هم از این الگو پیروی میکند. و در طول یک رشته بتا آنها بطور متناوب بالا و پایین صفحه بتا قرار میگیرند.
رشتههای بتا به دو طریق میتوانند برای تشکیل صفحات چین خورده با هم میانکنش داشته باشند یا به حالت صفحه موازی همسو[۳] و یا به حالت موازی ناهمسو[۴] مشاهده میشوند. چگونگی برقراری پیوندهای هیدروژنی در هر یک از این دو نوع، الگویی اختصاصی و قابل تشخیص دارد.
[۱] residue
[۲] Pleated
[۳] Parallel
[۴] anti parallel
۲۵۱-پیشگویی ساختار دوم و سوم پروتئین
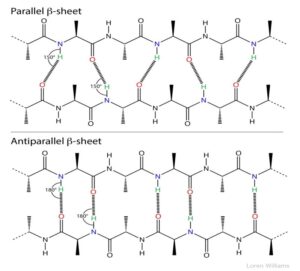
تصویر ۴-۱۲: نمایی از صفحات بتا موازی ناهمسو و همسو.
۳-۱-۱۲ سلسله مراتب ساختمانی پروتئینها
بیوشیمیست دانمارکی لانگ[۱] اصطلاحات ساختمان اول[۲]، دوم [۳]و سوم [۴]را برای تأکید بر سلسله مراتب ساختمانی در پروتئینها وضع کرد. ساختمان اول توالی اسیدهای آمینه یا به عبارت دیگر آرایش اسیدهای آمینه در یک زنجیره پلیپپتید خطی است. دو پروتئین مختلف که تشابه معنیداری در ساختمان اول داشته باشد گفته میشود که با یکدیگر همولوگ هستند و بنابراین توالی DNA آنها نیز مشابه است. باور عمومی بر این است که دو پروتئین همولوگ از نظر تکاملی نیز بهم مرتبط هستند و از یک ژن اجدادی مشترک تکامل یافتهاند.
ساختمان دوم بطور عمده از رشتههای بتا و مارپیچ آلفا ساخته شده است. تشکیل ساختمان دوم در یک منطقه محلی از زنجیره پلیپپتیدی تا حدی بوسیله ساختمان اول تعیین میشود. توالیهای آمینواسیدی خاصی برای رشتههای بتا یا مارپیچ آلفا مناسب است و بقیه برای تشکیل مناطق حلقه مناسب هستند. عناصر ساختمان دوم معمولاً خودشان را در شکل موتیفهای ساده آرایش میدهند. موتیفها بوسیله چفت شدن[۵] زنجیرههای جانبی مارپیچهای آلفا یا رشتههای بتای مجاور و نزدیک بهم تشکیل میشوند.معمولاً چندین موتیف برای تشکیل ساختارهای فشرده کروی [۶] – که دمین[۷] نامیده میشوند – با هم ترکیب میشوند.
اصطلاح ساختمان سوم را به عنوان یک اصطلاح مشترک هم برای طریقه آرایش موتیفها به ساختمانهای دمِین و هم برای راهی که یک زنجیره پلیپپتیدی به چندین دمِین تا[۸] میخورد بکار میبریم. در همه مواردی که مشاهده شده، نشان داده شده است که اگر توالی اسیدهای آمینه در دو دمِین در پروتئینهای مختلف همولوژی داشته باشند، این دمِینها ساختمان سوم مشابه دارند.
مولکولهای پروتئینی که فقط یک زنجیره اصلی دارند پروتئینهای تک رشته ای[۹] نامیده میشوند. اما تعداد زیادی از پروتئینها ساختمان چهارم [۱۰] دارند که از چند زنجیره پروتئین مشابه که در یک ملکول چند زنجیره ای [۱۱] تجمع یافتهاند
[۱] Kui linderstrom lang
[۲] primary
[۳] secondary
[۴] tertiary
[۵] packing
[۶] Compact globular
[۷] domain
[۸] fold
[۹] monomeric
[۱۰] quaternary
[۱۱] Multimeric
۲۵۲-فصل دوازدهم
تشکیل شدهاند. این زیرواحدها میتوانند بطور مستقل یا با اثر تعاونی روی یکدیگر بطوریکه فعالیت یک پروتئین وابسته به فعالیت زیر واحد دیگر باشد عمل کنند.
واحد بنیادی ساختمان سوم دمین است. یک دمین بعنوان یک زنجیره پلیپپتید یا قسمتی از یک زنجیره پلیپپتیدی تعریف شده است که میتواند بطور مستقل به ساختمان سوم پایدارتا بخورد. دمینها همچنین واحدهای فعالیت هستند. اغلب، به دمینهای مختلف یک پروتئین عملکردهای مختلف نسبت داده میشود.
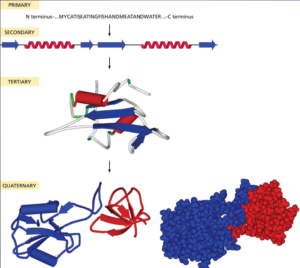
تصویر ۵-۱۲: سلسله مراتب ساختمانی پروتئینها.
۲-۱۲ روشهای تجربی تعیین ساختار پروتئین
کشف ساختار سوم و چهارم یک پروتئین راهنمای بسیار مهمی برای تعیین کارکرد این پروتئین است. از روشهای معمول میتوان به پراش اشعه ایکس و تشدید مغناطیسی هسته اشاره کرد. تاشدگی پروتئین فرایندی فیزیکی است که در آن پلیپپتید به ساختار سهبعدی مشخصی پیچیده میشود. هر پروتئین از یکی پلیپپتید آغاز میشود. پلیپپتید زنجیرهای از اسیدهای آمینه هستند که در آغاز هیچ ساختار سهبعدی مشخصی ندارند (سمت چپ شکل روبهرو). ولی هر اسید آمینه در زنجیره میتواند ویژگیهای شیمیایی خاصی داشته باشد؛ مثلاً آبدوست (هیدروفیل) یا آبگریز (هیدروفوب) یا باردار باشد. اسیدهای آمینه به خاطر این ویژگیها با یکدیگر و با محیط سلول برهمکنش میکنند و سرانجام ساختار سهبعدی ویژهای را که همان ساختار اصلی پروتئین است به خود میگیرند (سمت راست تصویر ۵-۱۲). این ساختار را ترتیب زنجیرهی اسیدهای آمینه تعیین میکنند. سازوکار تاشدگی پروتئین هنوز کاملاً شناخته نشده است.
اسیدهای آمینه ازیک گروه آمین و یک گروه کربوکسیل و یک زنجیرهی جانبی ساخته شدهاند وبا پیوندهای کوالانسی به هم متصلند. پیوند کوالانسی بین اسیدهای آمینه بسیار محکم است به طوری که امکان جابجایی و چرخیدن به اتمهای N-H و C=O را نمیدهد، اما سایر پیوندها دارای استحکام کمتری بوده و امکان چرخیدن برایشان وجود دارد. این ویژگی باعث میشود پروتئینها خاصیت انعطاف پذیری پیدا کنند و بتوانند تا بخورند.
۲۵۳-پیشگویی ساختار دوم و سوم پروتئین
پروتئینها در اثر تا خوردگی شکل فضایی را ایجاد میکنند که دارای کمترین انرژی است. وقتی پروتئین تا میخورد پیوندهای ضعیف غیر کوالانسی به تثبیت تا خوردگی کمک میکنند. این پیوندها عبارتند از: پیوند یونی، پیوند هیدروژنی، برهمکنشهای واندروالس و نیروهای آب گریز. همانطور که گفته شد زنجیره جانبی متصل به اسیدهای آمینه میتوانند قطبی یا غیر قطبی باشند. وقتی پروتئین در آب سیتوزولی قرار میگیرد زنجیرههای جانبی آب گریز تمایل دارند بصورت یک مجموعه در درون مولکول پروتئین قرار بگیرند تا حداقل تماس با مولکولهای قطبی آب را داشته باشند و اثر تضعیف کننده آنها بر پیوندهای هیدروژنی آب حداقل باشد. از طرف دیگر زنجیرههای جانبیای که قطبی هستند تمایل دارند که در روی سطح مولکول پروتئین قرار بگیرند و با مولکولهای آب و یا سایر مولکولها پیوند هیدروژنی ایجاد کنند. بنابراین خاصیت آب گریزی زنجیرههای جانبی در تعیین شکل پروتئین نقش اساسی دارد.
در اثر تا خوردن، پروتئینها شکل نهایی پیدا میکنند که یکتا و دارای کمینه انرژی است. تمامی اطلاعات لازم برای تا خوردن پروتئین در توالی اسیدهای آمینه آن نهفتهاست. زیرا این توالی منحصر به فرد محل قرارگیری زنجیرهای جانبی را تعیین کرده وشکل نهایی پروتئین را تعیین میکنند. تا خوردن پروتئین در درون سلول یکی از فرایندهای حیاتی زیستی است. در اثرتا خوردن نامناسب تجمعاتی ایجاد میشود که میتواند به بافت سلول آسیب برساند. برخی از بیماریهای عصبی مانند آلزایمر و بیماری هانتینگتون در اثر بد تا خوردن پروتئین در بافتهای عصبی ایجاد میشوند. در فرایند تا خوردن، پروتئین مسیرهایی را از نظر سطح انرژی طی میکند. در ابتدا پروتئین یک رشتهی فاقد ساختار مشخص با انتروپی بالاست. ضمن فرایند تا خوردن اتمهای ستون فقرات پروتئینی با ایجاد انواع پیوندهای مناسب یا نا مناسب با سایر اتمهای ستون فقرات پروتئینی ویا زنجیره جانبی باعث تغییر در سطح انرژی پروتئین میشوند.
فرایند تا خوردن که درآن پروتئین از حالت تا نخورده به حالت نهایی میرسد را میتوان از دیدگاه آماری بررسی نمود. میتوان به جای یک پروتئین که فرایند تا خوردن را با تعداد زیادی پیکربندی ممکن از نظر انرژی تجربه میکند یک مجموعه از همه پیکر بندیهای ممکن انرژی در نظر گرفت و تعداد حالتهایی که در آن انرژی مقدار خاصی است را بدست آورد و برای آن سیستم آنتروپی را محاسبه نمود و به این ترتیب انرژی آزاد گیبس را به دست آورد. حالت نهایی سیستم در کمینه انرژی اتفاق میافتد. بررسی سیستم از نظر اماری به ما این امکان را میدهد که فرایند تا خوردن را با جزئیات شبیه سازی کنیم و این فرایند را بهتر بشناسیم.
پارادوکس لوینتال
یکی از مباحث جالب درتا خوردن پروتئین، مسئله زمان تا خوردن است. مدت زمان لازم برای این فرایند در درون سلول از مرتبه ثانیهاست. نخستین بار لوینتال با محاسبه پارامتر سرعت واکنش از رابطه آرنیوس مدت زمان لازم برای تا خوردن پروتئین را محاسبه کرد. وی این زمان را با در نظر گرفتن تمامی مسیرهای ممکن انرژی انجام داد و جوابی که بدست آورد یک عدد نجومی بود. لوینتال نتیجه گرفت که در فرایند تا خوردن پروتئین، همه مسیرهای ممکن انرژی طی نمیشوند و مسیر ویژهای برای تا خوردن وجود دارد.
تعیین ساختار سهبعدی پروتئین به طور تجربی بسیار دشوار است. ولی در مورد ترتیب زنجیرهی توالیهای هر پروتئین معمولاً اطلاعات موجود است. پژوهشگران میکوشند روشهای زیستفیزیکی گوناگون را به کار بگیرند تا ساختار سهبعدی نهایی را از روی دنبالهی اسیدهای آمینه پیشبینی کنند. بسیاری از پروتئینها برای کار حتماً باید ساختار سهبعدی درست خود را دارا باشند. معمولاً اگر پروتئینی نتواند به ساختار درست خود تا شود، غیرفعال میشود.
۳-۱۲ پیشگویی ساختمان پروتئینها
داشتن اطلاعات درباره جزئیات ساختمان سه بعدی یک پروتئین برای توسعه درک عملکرد آن ضروری است. متاسفانه در مقایسه با تعداد بسیار زیاد توالیهای شناخته شده، ساختمان تعداد کمی از پروتئینها شناخته شده است. با رشد بیشتر شکاف بین توالیها و ساختمانهای شناخته شده، نیاز به توسعه روشهای پیشگویی ساختمان که قابل اعتماد باشند افزایش یافتهاست. بویژه برای پروتئینهایی که تعیین ساختمان آنها بطریق تجربی ممکن نباشد و پروتئینی با توالی مشابه با آن که ساختمانش شناخته شده باشد نیز یافت نشود. بعلاوه روشهای پیشگویی موفق، به روشن شدن رابطه بین توالی اسیدهای آمینه و ساختمان پروتئینها کمک میکند و میتواند به طراحی پروتئینهای جدید و مهندسی آنها کمک کند.
۲۵۴-فصل دوازدهم
۱-۳-۱۲ طبقه بندی ساختمانهای پروتئینی
رشد روز افزون ساختمان پروتئینهای شناخته شده،یافتن یک شمای کلی از ساختمانهای مختلف، یافتن مشابهتها و همینطور به منظور بهترشدن روشهای پیش گویی بهترین راهکار طبقه بندی ساختمانهای پروتئینی میباشد مثلا براساس یک توجه ساده به موتیفهای متصل بهم، Michael Levitt و Cyrus Chotia یک طبقهبندی از ساختمانهای پروتئینی استخراج کرده و ساختمان دمینها را به سه گروه اصلی طبقهبندی کردند. a دمینها، bدمینها وa/b دمینها. در ساختمانهای a، هسته [۱]بطور انحصاری از مارپیچهای a ساخته شده است. در ساختمانهای b هسته از صفحات b موازی ناهمسو تشکیل شده و معمولاً دو صفحه b برروی یکدیگر چفت[۲] شدهاند. ساختمانهای a/b از ترکیبی از موتیفهای b-α-b ساخته شدهاند که یک صفحه b موازی که غالباً همسو میباشد بهوسیلهی مارپیچهای a احاطه شده است.
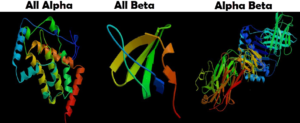
تصویر ۶-۱۲: طبقه بندی ساختمانهای پروتئینی.
بعضی از پروتئینها از ترکیبی از موتیفهای b و α جدا از هم ساخته میشوند و معمولاً این ترکیبات یک صفحه کوچک b موازی ناهمسو در یک قسمت از دمِین که در برابر تعدادی از مارپیچهای α چفت شده است تشکیل میدهند که این ساختمانها را میتوان متعلق به چهارمین گروه کوچکی بنام b+ α در نظر گرفت. علاوه براین گروهها، تعدادی از پروتئینهای کوچک هستند که غنی از پیوندهای دیسولفیدی یا اتمهای فلزی هستند و یک گروه خاص را تشکیل میدهند. به نظر میرسد ساختمان این پروتئینها از حضوراین فلزات یا دیسولفیدها به شدت تأثیر میگیرد و اغلب همانند نسخههای به هم ریخته ای[۳] (غیرطبیعی) از پروتئینهای عادی تر بنظر میرسند.
طبقه بندیها یا همان کلاسیفیکیشنهای متعددی برای پروتئینها وجود دارد که هر کدام از این طبقهبندیها بر اساس یک اصولی بنا شده است. مثلا بانک اطلاعاتی SCOP بر اساس چشم محقق استوار است و ساختمانها را طبقه بندی نموده و ارتباط تکاملی و ساختمانی را نمایش میدهد. SCOP توسط Murzin و همکارانش در سال ۱۹۹۵ ایجاد شد. سطوح طبقهبندی در SCOP به شرح CLASS, FOLD, SUPERFAMILY, FAMILY میباشد که در تصویر ۷-۱۲ نمایش داده شده است.
[۱] Core
[۲] pack
[۳] distorted version
۲۵۵-پیشگویی ساختار دوم و سوم پروتئین
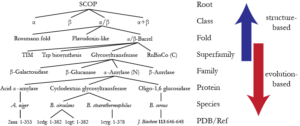
تصویر ۷-۱۲: کلاسیفیکیشن بانک اطلاعاتی SCOP .
با توجه به این که طبقه بندیهای مختلفی برای پروتئینها وجود دارد در جدول ۱-۱۲ تعدادی از مهمترین بانکها و نحوه طبقه بندی آنها و شرحی در رابطه با انواع طبقه بندیها آورده شده است.
جدول ۱-۱۲: انواع طبقه بندی پروتئینها.
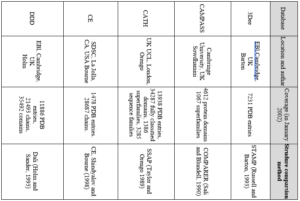
۲۵۶-فصل دوازدهم
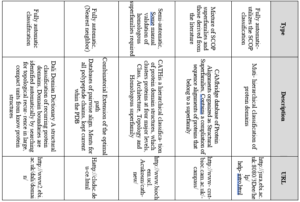
ادامه جدول ۱-۱۲: انواع طبقه بندی پروتئینها.
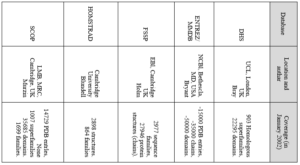
۲۵۷-پیشگویی ساختار دوم و سوم پروتئین
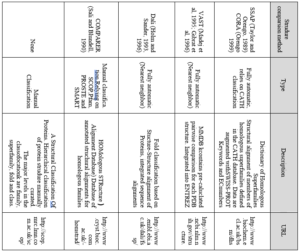
دلایل پیشگویی کلاس ساختاری پروتئینها
در طی دهه اخیر پیشگویی کلاس ساختاری پروتئینها با روشهای گوناگون به دلایل زیر بسیار مورد توجه بوده است:
۱ – مفاهیم کلاس ساختاری پروتئینها بر اساس محتوای ساختار دوم در یک پروتئین از نقطه نظر تجربی و تئوری بسیار مفید میباشد.
۲ – کلاس ساختاری یک پروتئین بیان کننده ساختار کلی آن پروتئین است که میتواند در بهبود مطالعات ساختمانی پروتئین موثر باشد.
۳ – با داشتن کلاس ساختاری پروتئینها میتوان دقت پیشگویی ساختار دوم آنها را از روی ردیف اسیدهای آمینه بطور قابل توجهی بهبود بخشید.
۴ – با اطلاعات بدست آمده از کلاس ساختاری پروتئین میتوان تعداد ساختارهای احتمالی را در روش کمینه کردن انرژی جهت تعیین ساختار سوم یک پروتئین محدود کرد.
۵ – علاوه بر نقش مهم کلاس ساختاری پروتئین در بهبود محاسبات ضرایب آبگریز (تعیین سطح در دسترس پروتئین)، میتوان ارتباط آن را با خواص گوناگون پروتئین مثل قرار گرفتن در اجزاء خارج یا داخل سلولی و یا فعالیت زیستی (آنزیم بودن یا نبودن) ذکر کرد.
۲-۳-۱۲ روشهای پیشگویی ساختار دوم
بیش از بیست روش مختلف برای پیشگویی ساختار دوم وجود دارد. اساس این روشها بر این است که، توالیها موضعی (مثل ساختمان دوم) هستند. این روشها از کارآیی بالا برخوردار نیستند. برخی از این روشها عبارتند از:
- Chou- Fasman and GOR methods
- NN models
- Nearest neighbor Methods
- HMMs
۲۵۸-فصل دوازدهم
۱-۲-۳-۱۲ روش چو- فاسمن
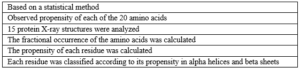
این روش مبتنی بر محاسبه پارامترهای پیکربندی از روی فراوانیهای مشاهده شده اسیدهای آمینه در پروتئینها با ساختمان تعیین شده میباشد. در این روش به عنوان پارامتر کنفورماسیونی اسیدهای آمینه iام برای پیکربندی x به صورت زیر تعریف میشود.
تعداد مشاهده شده اسید آمینه iام در حالت پیکربندی x، تعداد کل اسیدهای آمینه iام، تعدا مشاهده شده اسیدهای آمینه در حالت پیکربندی X و N تعداد کل اسیدهای آمینه بانک اطلاعاتی میباشد. این الگوریتم بر اساس جستوجوی هستههایی با طول ۴ تا ۶ اسید آمینه و سپس رشد آن از دو طرف مبتنی است. دقت این روش در حدود ۵۱ درصد است.
جدول ۲-۱۲: خصوصیات روش پیشگویی Chou-fasman.
۲-۲-۳-۱۲ روش گور
در این روش، رشتهای از اسیدهای آمینه یک زنجیره پلیپپتیدی را به عنوان پیغامی که به وسیله فرآیند تاشدگی به پیغام دیگری (رشتهای از حالتهای پیکربندی) ترجمه میشود در نظر گرفته میشود. این روش بر اساس تئوری اطلاعات به روش پیشگویی گور معروف است. دقت این روش ۶۳ درصد است.
۳-۲-۳-۱۲ روشهای تشابه ترکیب
اساس این روش آن است که پپتیدهای مشابه دارای ساختمانهای دوم یکسانی هستند. این روشها از لحاظ تعریف و نمرهدهی تشابه، متفاوت هستند. این روشها از طول ۱۱ تا ۱۷ اسید آمینهای استفاده میکنند و پیشگویی آنها بر اساس بیشترین نمره شباهت میباشد. به کمک مقیاس اطمینان است. صحت این روش ۲۱درصد است.
۴-۲-۳-۱۲ شبکههای عصبی و روش شناسایی الگوها
در این روش به اسیدآمینه مربوط به توالی، کدی داده میشود که لایه ورودی را میسازند. ورودیها در یک لایه معروف به لایه پنهان در یک وزن یا فاکتور اتصال به ازاء هر حالت پیکربندی ضرب میشوند و یک فاکتور جدید به عنوان خروجی را ارائه میدهد و خروجیها تفسیر میشوند. دقت این روش ۵۰ تا ۶۰ درصد است.
پایگاههای بسیاری به منظور پیشگویی ساختار دوم پروتئینها موجود میباشند که برخی از آنها در جدول ۳-۱۲ آمده است. بسیاری از پایگاهها به علت حجم زیاد مراجعات توالی را از کاربر دریافت میکنند و بعد از مدتی نتیجه را از طریق ایمیل برای کاربر ارسال میکنند.
۲۵۹-پیشگویی ساختار دوم و سوم پروتئین
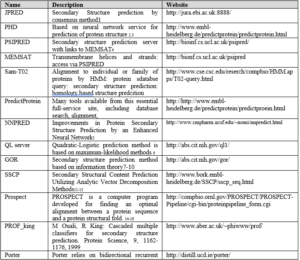
جدول ۳-۱۲: پایگاههایی به منظور پیشگویی ساختار دوم پروتئین.

پایگاههای بسیاری به منظور پیشگویی ساختار دوم پروتئینها موجود میباشند که برخی از آنها در جدول ۳-۱۲ آمده است. بسیاری از پایگاهها به علت حجم زیاد مراجعات توالی را از کاربر دریافت میکنند و بعد از مدتی نتیجه را از طریق ایمیل برای کاربر ارسال میکنند.
جدول ۳-۱۲: پایگاههایی به منظور پیشگویی ساختار دوم پروتئین.
۲۶۰-فصل دوازدهم
۳-۳-۱۲ روشهای پیشگویی ساختار سوم
رشد سریع اطلاعات مربوط به توالیهای پروتیئنی در مقایسه با ساختارهای پروتئینی، بیانگر نیاز روزافزون به روشهای دیگری در تعیین ساختار سوم پروتئینها است. تعیین ساختار سوم پروتئین با روشهای تجربی وقتگیر، پرهزینه و در مواردی غیرممکن است. استفاده از روشهای تئوری پیشگویی ساختار که تنها با کمک توالی یا ساختار اولیه پروتئین اقدام به محاسبه و پیشگویی ساختار پروتئین میکند، یکی از ابزارهای نوین در مطالعه پروتئینهاست.
پیشگویی ساختار سه بعدی پروتئینها براساس سه روش اصلی انجام میشود:
۱-۳-۳-۱۲ مدل سازی همولوژیکی
اساس پیشبینی توسط مدل سازی همولوژیکی این است که توالی پروتئین با یک یا تعداد بیشتر پروتئین با ساختار شناخته شده شباهت داشته باشد. این روش، مدل سازی مقایسهای (CA) نیز نامیده میشود بر اساس این واقعیت که پروتئینهایی با توالیهای مشابه، ساختارهای مشابه دارند. این روش بر آن اساس که، پروتئینهای یک خانواده، بیشتر توالیهای آمینو اسیدی شان حفاظت شده است، آسان میباشد. چرا که وقتی ساختار یک پروتئین از یک خانواده به وسیلهی آزمایش تعیین شود، اعضای دیگر آن خانواده میتوانند بر اساس تطابق شان با ساختار شناخته شده مدل سازی شوند. دقت پیش گویی با این روش به میزان تشابه بین توالی پروتئین هدف و ساختارهای الگو بستگی دارد. مشکل عمده مدل سازی همولوژیکی پیدا کردن توالی هدفی است که به عنوان الگو استفاده میشود. تقریبا ۵۷% همهی توالیهای شناخته شده حداقل یک دمین دارند که به یک پروتئین با ساختار شناخته شده وابسته میباشد.
بر اساس اصول فوق، منطقی است که پروتئینهای هومولوگ ساختارهای بسیار شبیه به هم را به خود بگیرند. از آنجا که یک فولد پروتئین از نظر تکاملی بسیار حفظ شدهتر از توالی آمینو اسیدی آن است، یک توالی هدف میتواند با دقت و صحت بالایی با توجه به یک الگو مربوط که از روی تطبیق توالیهای آمینواسیدی آنها با هم به دست آمده است، مدلسازی شود. چنانچه الگو و هدف، توالیهای مشابهی داشته باشند، همولوژی مدلینگ بسیار دقیق خواهد بود.
۲۶۱-پیشگویی ساختار دوم و سوم پروتئین
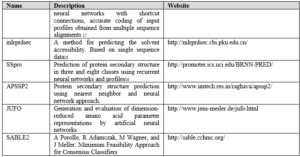
جدول ۴-۱۲: معرفی برخی از سرورهای مدل سازی همولوژیکی.

۲-۳-۳-۱۲ تکنیک AB initio
در این روش سعی میشود تا مدل ساختار سه بعدی پروتئین تنها با استفاده از توالی و بررسی نیروها ایجاد گردد و از اطلاعات ساختاری موجود استفاده نمیشود. AB initio method، همچنین روش De-novo (مدلسازی ابتدا به ساکن) و یا
Free modeling techniques نامیده میشود. اصطلاح AB initio method در ابتدا اشاره به روشهایی داشت که برای پیشبینی ساختار پروتئین انجام میشد و از دانستههای آزمایشگاهی مربوط به ساختار استفاده نمیکرد. بعدها این اصطلاح با به وجود آمدن روشهایی بر اساس فراگمنتها مبهم شد، چرا که این روشها براساس این واقعیت به کار میروند که گر چه ما نمیتوانیم همهی فولدهای استفاده شده در بیولوژی را ببینیم، احتمالا قادر خواهیم بود تقریبا همهی زیر ساختارها را ببینیم. در این روش فرض میشود که کمترین انرژی آزاد در طول عمر پروتئین میبایست در دسترس باشد و تلاش میشود تا این مقدار کم را به وسیله بررسی بسیاری از کانفورماسیونهای ممکن پروتئین به دست آورند.
AB initio method به دو کلاس عمده تقسیم میشود:
الف) AB initio method به وسیلهی اطلاعات پایگاه داده
ب) AB initio method بدون اطلاعات پایگاه داده
اگر چه روشهای این گروه از لحاظ کامپیوتری بسیار پیچیده و هنوزفاقد دقت هستند، به چند دلیل به طور مداوم استفاده میشوند و گسترش مییابند. اولا این که در برخی از موارد حتی یک ساختار همولوگ وابسته خیلی دورهم ممکن است در دسترس نباشد. در این موارد ab initio تنها روش میباشد. دوما این که ممکن است ساختارهای جدیدی کشف شوند که به وسیلهی روشهایی که اساس شان مقایسهی ساختارهای شناخته شده است قابل شناسایی نباشند.
بر اساس مدل AB initio شکل فضایی مولکول به گونهای است که به حداقل سطح انرژی برسد (استفاده از قوانین شیمی، فیزیک و ترمودینامیک). روشهای ابتدا به ساکن یا de novo- به دنبال ساختن مدلهای سه بعدی برای پروتئین از روی توالی آن، بر اساس اصول فیزیکی به جای استفاده از ساختارهای تعیین شده قبلی هستند. برای شبیهسازی فرآیند تا خوردن پروتئینها روشهای گوناگون وجود دارند از جمله روشهای استوکستیک که برای جستوجوی راه حل، به عنوان نمونه global optimization یک پارامتر انرژی مناسب را به کار میبرند.
۲۶۲-فصل دوازدهم
۳-۳-۳-۱۲Threading و تشخیص Fold
این روشfold recognition و۳D- 1D flod recognition نیز نامیده میشود. همچنین در بعضی منابع «بندکش» ترجمه شده است. این روش یک رویکرد حد واسط دو روش قبلی است که هم از تشابه توالی در صورت وجود و هم از اطلاعات مربوط به تطابقهای ساختاری استفاده میکند. هدف این روش تطابق توالی هدف با ساختار شناخته شده در کتابخانهای از فولدها میباشد. در این روش توالی آمینواسیدی پروتئینی که ساختار آن شناخته شده نیست، بر علیه یک بانک اطلاعاتی از ساختارهای تعیین شده پویش میشود و در هر مورد، یک پارامتر امتیازدهی برای تعیین میزان همخوانی توالی با ساختار استفاده میشود و مدلهای سه بعدی محتمل پیشنهاد میگردد.
در مقایسه با میلیونها توالی پروتئین موجود تنها تعداد کمی Protoin Folds موجود است. این بدین معناست که ساختارهای پروتئین از توالی پروتئین محافظت شدهتر هستند. در نتیجه بسیاری از پروتئینها میتوانند بدون اینکه مشابهت توالی داشته باشند، پینخوردگی مشابهی داشته باشند.
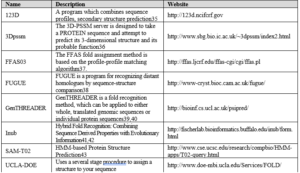
جدول ۵-۱۲: معرفی برخی از سرورهای متد fold recognition
به دلیل طرفداران زیاد پیشبینی ساختار پروتئین به وسیلهی روشهای کامپیوتری، جوامع علمی تلاشهای زیادی را در حل مشکلات مختلف و محدودیتهای هر کدام از این روشها دارند. بر همین اساس با توجه به تصویر ۸-۱۲ میبینید یک روند کلی برای پیشگویی ساختار سوم پروتئینها تدوین شده است.
۲۶۳-پیشگویی ساختار دوم و سوم پروتئین
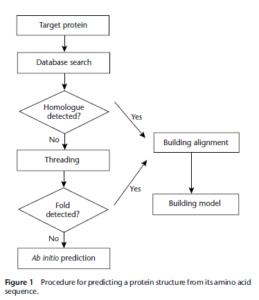
تصویر ۸-۱۲: روند کلی پیشبینی ساختار پروتئین.
در رابطه با Threading و تشخیص فولد الگوریتمها میتوانند در دو گروه قرار گیرند. الگوریتمهای Pairwice energy (Threading) و الگوریتمهای Profile (Foldrecognition). این الگورتیمها بهترتیب تحت عنوان مدل سازی مقایسهای و پیشگویی ساختمان سوم به وسیله کنار هم قرار دادن عناصر ساختمان دوم نیز شناخته میشوند که در زیر شرح داده شدهاند.
۴-۳-۳-۱۲ مدلسازی مقایسهای
از تشابه توالی پلیپپتیدهایی با ساختمان (ساختار نه توالی) مشابه استفاده میشود. بر این اساس پروتئینها با توالی مشابه، شکل فضایی شبیه به هم پیدا میکنند. این روش، از ساختارهای از قبل تعیین شده به عنوان نقطه شروع و یا الگو استفاده میکند. اساس این روش را knowledge- based میدانند یعنی بر اساس اطلاعات ساختاری موجود در ساختار یک پروتئین طبیعی که به روشهای تجربی تعیین ساختار شده است، ساختاری برای پروتئین مورد نظر پیشگویی میشود. در این روش برای رسیدن به کمینه سطح انرژی آزاد یک پروتئین با ساختار ناشناخته و توالی معلوم، از نمونه طبیعی که از نظر توالی با آن پروتئین شباهت قابل قبولی داشته باشد استفاده میشود. این الگوریتم Pairwise energy نیز نامیده میشوند.
۵-۳-۳-۱۲ پیشگویی ساختمان سوم به وسیله در کنار هم قرار دادن عناصر ساختمان دوم (profile Method)
در این روش شکل تقریبی ساختمان سوم از کنار هم قرار دادن ساختمانهای دوم به طریق مختلف ایجاد میشود که با قرار دادن شرط نهایی، فقط برخی از این ساختمانها پذیرفته میشوند. در این روش از عبارات کمی که در مورد نیروهای مختلف موجود در ساختارهای پروتئینی بیان گردیده است باید انرژی پتانسیل کل یک پروتئین در یک کانفیگوراسیون مشخص محاسبه شود. در حقیقت تلاش اصلی، یافتن یک عبارت خوب برای انرژی پتانسیل یک پروتئین به صورت تابعی از موقعیت همه اتمهای آن است.
در این روش یک پروفایل برای گروهی از ساختارهای پروتئینی مرتبط ساخته میشود. پروفایل ساختاری با منطبقسازی ساختارها برای باقیماندههای متناظر تولید میشود. سپس از اطلاعات آماری این باقیماندههای انطباق یافته برای ساخت پروفایل استفاده میگردد. پروفایل شامل امتیازهایی است که تمایل هر یک از بیست اسیدآمینه را برای قرار گرفتن در هر یک از موقعیتهای پروفایل توصیف میکند. امتیازهای پروفایل شامل اطلاعات انواع ساختارهای دوم، میزان قرارگیری در معرض حلال، قطبیت و آبگریزی اسیدآمینهها است. برای پیشگویی فولد ساختاری یک توالی ناشناخته، توالی درخواستی ابتدا برای حضور ساختارهای دوم، دسترسی به حلال و قطبیت پیشگویی میشود. سپس از اطلاعات پیشگویی شده برای
۲۶۴-فصل دوازدهم
مقایسه با پروفایلهای تمایلی فولدهای ساختاری مشخص، برای یافتن فولدی که بهترین پروفایل پیشگویی شده را ارائه میدهد، استفاده میشود.
به دلیل آنکه Threading و تشخیص فولد، هومولوگهای ساختاری را بدون آنکه کاملاً به شباهتهای توالی وابسته باشند، تشخیص میدهند، نشان داده شده است که نسبت به PSI-BLAST حساسیت بیشتری را در یافتن روابط تکاملی دور دارا میباشند.
۴-۳-۱۲ ارزیابی کیفیت پارامترهای ساختاری مدل ساخته شده
معمولاً بعد از پیشگویی ساختار سوم یک پروتئین بهمنظور ارزیابی کیفیت پارامترهای ساختاری مدل ساخته شده نمودار راماچاندرا برای الگو و پروتئین هدف ترسیم میشود. نمودار راماچاندران وضعیت مجاز هر زاویه را برای ساختارهای پروتئین را نشان میدهد و با نامهای Ramachandran plot یا plot ψ] و [φ شناخته میشود.
هر گاه محور Xها با φ و محور Y با ψ مشخص میشود و مقادیر ψ و φ هر کدام از اسیدهای آمینه شرکت کننده در ساختار پروتئینها را مشخص کنیم، نقشهای ایجاد میشود که بهنام نقشه ساختمان فضایی یا نمودار راماچاندران معروف است. بهعبارتی هر اسید آمینهای مجموعه از مقادیر این دو زاویه را به خود اختصاص میدهد و هیچ دو اسید آمینهای نقش دقیقاً یکسانی ندارند و مثل اثر انگشت در انسان خاص همان اسید آمینه هستند.
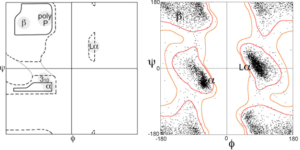
تصویر ۹-۱۲: تصویری از نمودار راماچاندرا.
۴-۱۲ داکینگ مولکولی
تکنیک محاسباتی میباشد که میتواند برهمکنش بین دو مولکول را پیشگویی کند. این تکنیک بهطور عمده شامل الگوریتمهایی مانند دینامیک مولکولی، شیبهسازی مونت کارلوو، روش جستوجو براساس بررسی قطعات و … میباشد. مطالعات داکینگ مولکولی در تعیین بر همکنش بین دو مولکول، برای پیدا کردن بهترین جهتگیری یک لیگاند در یک کمپلکس با حداقل انرژی بهکار برده میشود نتایج بهدست آمده توسط یک تابع درجهبندی آماری تجزیه و تحلیل میشود، این تابع درجهبندی آماری برای محاسبه انرژی انترکشن آن را به مقادیر عددی به نام درجه داکینگ تبدیل میکند. نتایج بهدست آمده از داکینگ شامل اشکال ۳ بعدی از لیگاند متصل شده به ماکرومولکول میباشد که میتوان با ابزارهای مشاهدهگر مانند pymol و rasmol مشاهده کرد و میتوان در بهدست آوردن بهترین حالت از لیگاند برای اینترکشن با ماکرومولکول به ما کمک کند.
فرآیند داکینگ شامل سه فاز اصلی است. فاز یک، مرحله نمونهگیری، شامل ایجاد کانفورماسیونهای مختلف لیگاند و بررسی جهتگیری آنها نسبت به جایگاه فعال رسپتور است. وقتی انعطافپذیری رسپتور هم مد نظر باشد، فاز نمونهگیری شامل تغییرات کانفورماسیونی رسپتور نیز میشود. در فاز دوم، مرحله امتیازدهی، تمایل اتصل لیگاند به رسپتور تخمین زده میشود. وقتی که داکینگ در جستوجوی مجازی ترکیبات یک کتابخانه مجازی مورد استفاده قرار میگیرد، ترکیبات براساس حالت با بهترین امتیاز رتبهبندی میشوند. امتیاز اختصاص یافته به هر حالت بر اساس ارزیابی تابع امتیازدهی که اغلب انرژی آزاد اتصال لیگاند – رسپتور را نشان میدهد بهدست میآید. در اکثر موارد
۲۶۵-پیشگویی ساختار دوم و سوم پروتئین
نرمافزارهای داکینگ سادهترین حالت نمایش رسپتور را با صرفنظر از انعطافپذیری آن و همچنین نادیده گرفتن مولکولهای حلال با هدف کاهش حجم محاسبات بهکار میبرند.
یکی از روشهای اندازهگیری کارایی الگوریتم داکینگ مولکولی، بررسی قدرت نرمافزار در پیشبینی نحوه اتصال لیگاند کریستالوگرافی به همان رسپتور است که از طریق تعریف ریشه میانگین مربعات انحراف از حالت گریستالوگرافی اندازهگیری میشود. اگرچه کیفیت دستهبندی براساس RMSD در مورد مولکولهای کوچک و بزرگ مشکلساز است، این روش بهطور گسترده به عنوان معیاری برای تعریف موفقیت با شکست الگوریتم داکینگ بهکاربرده میشود. معیار دوم برای سنجش کارایی الگوریتم داکینگ مولکولی توانایی آن در پیشبینی تمایل لیگاندهای مختلف (انرژی آزاد پیوند شدن) است. چون مقیاس امتیازدهی داکینگ معمولاً در محدوده دادههای تجربی نیست، اغلب ارتباط بین امتیاز داکینگ xi و دادههای تجربی yi با استفاده از ضریب ارتباطی پیرسون CP بیان میشود. روش دیگر برای ارزیابی نتایج جستوجوی مجازی، منحنی مشخصه عملکرد سیستم یا منحنی عملیاتی دریافت کننده (ROC) است که بهطور گسترده در زمینههای مختلف بهکار برده میشود.
همانطورکه در بالا توضیح داده شد مرحله آخر داکینگ مولکول آنالیز نتایج شامل محاسبه انرژی پیوند، Kd و RMSD میباشد. خطای جذر میانگین مربعات یا انحراف جذر میانگین ((RMSD) root- mean- square deviation) یا
((RMSE) root – mean- square error) تفاوت میان مقدار پیشبینی شده توسط مدل یا برآوردگر آماری و مقدار واقعی میباشد. RMSD یک ابزار خوبی است برای مقایسه خطاهای پیشبینی توسط یک مجموعه داده است و برای مقایسه چند مجموعه داده کاربرد ندارد. همچنین این تفاوتهای مجزا را ماندهها مینامند و خطای جذر میانگین مربعات برای جمعآوری آنها در یک عدد کاربرد دارد.
شیب نمودار RMSD بیان کننده پایدار بودن مدل در طول زمان شیبهسازی (۱۰ نانو ثانیه) است. هر چه این شیب به صفر نزدیکتر باشد مدل شبیهسازی شده پایدارتر و هر چه شیب به تدریج افزایش یابد و یا نوسان زیادی داشته باشد مدل، ناپدارتر خواهد بود. در رابطه با نموداری که در تصویر ۱۰-۱۲ مشاهده میکنید هیچ ناپایداری در زمان شبیهسازی مشاهده نشده است.
نمودار دیگری که روند صحیح فرآیند شبیهسازی را نشان میدهد نمودار انرژی است که در تصویر ۱۱-۱۲ مشاهده میکنید. برخلاف نمودار RMSD کاهش شیب نمودار انرژی بیانگر کیفیت بهتر شبیهسازی است چرا که انرژی کمتر در حالت پایدار بیشتر حاصل میشود. تغییرات انرژی در این نمودار پس از کاهش در ابتدای فرآیند در باقی مسیر یا نوسان ثابتی همراه بوده و بنابراین مدل ساخته شده در طول زمان شبیهسازی پایدار میباشد. بازه نوسان به مقدار ۰۰۲/۰ تا ۰۰۳/۰ کیلوژول بر مول قابل قبول است. در نمودار انرژی که در تصویر ۱۱-۱۲ مشاهده میکنید بازه نوسان کمتر از مقداری است که اشاره شد.
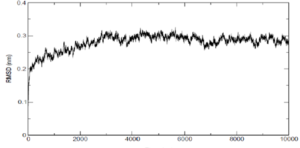
تصویر ۱۰-۱۲: نمودار RMSD برای مدلهای ساخته شده در GROMACS.
۲۶۶-فصل دوازدهم
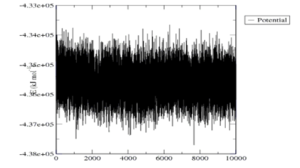
تصویر ۱۱-۱۲: نمودار انرژی پروتئین شبیهسازی شده در طول ۱۰ نانو ثانیه.